
*Paramyotonia Congenita
Physician’s Summary
Paramyotonia Congenita (PMC) is one of the periodic paralyses caused by mutations in the sodium channel. PMC causes muscle stiffness (myotonia) which is made worse by chilling or activity. Myotonia usually eases when the patients moves about, or “warms up” through physical activity. In PMC myotonia develops during activity, which is paradoxical or self-contradictory. This is where Paramyotonia Congenita gets its name, from the paradoxical nature of the myotonia.
Patients also have attacks of weakness and/or paralysis, alone or following an attack of paramyotonia. The patient’s serum potassium level during attacks may be low, normal or high. Patients who have PMC in combination with Hyperkalemic Periodic Paralysis may have muscles which appear very well-developed and strong, along with the progressive muscle weakness that occurs in HyperKPP. PMC is inherited from one parent who has the genetic mutation.
Symptoms often begin in infancy but are always present by the time the patient is a teenager. Almost everyone who inherits the mutation has symptoms, there are few unaffected “carriers”. Patients may move stiffly or appear ‘tense’. Some describe problems letting go of objects like pens, doorknobs or beverage containers. Strong voluntary contraction also may be associated with a long-lasting decrease in strength, which is not clearly due to an increase in muscle stiffness. Physical exertion such as carrying a heavy object, pushing a stalled car or lifting weights can trigger weakness which takes weeks (even months) to resolve. Patients with severe PMC may experience shortness of breath during attacks due to tightness of chest muscles.
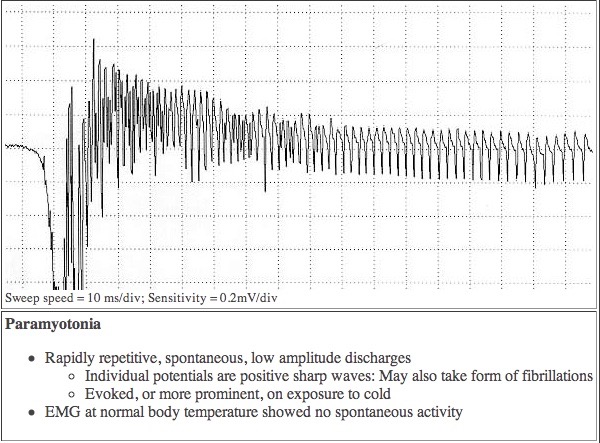
PMC was the first disorder recognized to include temperature-sensitive mutations in humans. At normal temperatures, there may not be any symptoms, but even a minor drop in temperature is enough to cause a malfunction which leads to muscle stiffness and/or weakness. This can be seen in the EMG tracing above.
Chilling first causes uncontrollable muscle tension. This tension is due to the muscle fibers rapidly contracting many times, rather than just once, each time they receive the signal to move. When the muscle is thoroughly chilled, the stiffness disappears and the muscle becomes flaccid (rag-doll-like) and paralyzed. The weakness may far outlast the exposure to cold. Even eating ice cream or swimming in cold water can be dangerous to those with severe cases of PMC.
In some families affected members have classical symptoms of PMC (muscle rigidity and weakness precipitated by cold and activity) without any other symptoms. But in other families affected members have attacks of both PMC and HyperKPP. The occurrence of both sets of symptoms in severe form was termed paralysis periodica paramyotonica (PPP); however, a continuum seems to exist, with PPP families and families having ‘pure’ PMC presenting the two ends of the spectrum.
While it has generally been reported that symptoms do not get worse with age and there is no muscle wasting, some patient’s symptoms do increase in the same way as attacks in patients with Hypokalemic Periodic Paralysis. That is to say, attacks become less acute but last longer, feeling in effect like deep fatigue which does not lift for weeks to months. In Hypokalemic Periodic Paralysis these attacks are called “abortive attacks“. Older patients with PMC also report the development of permanent muscle weakness which does not respond to medication.
In the Paramyotonias the sodium channels located in the skeletal muscle membrane fails to regulate the flow of sodium ions properly. The ratios of sodium and potassium inside and outside the cell become unbalanced. At first this imbalance causes the muscle fiber to contract inappropriately and repeatedly, but as the imbalance worsens the muscle stops responding to nerve signals and becomes weak or paralyzed. This can clearly be seen in an EMG measurement of muscle from a PMC patient.
The mutations for Paramyotonia Congenita and the Paramyotonia Syndromes are located in voltage-gated sodium channel SCN4A, which is on chromosome 17. These mutations cause so-called “gating” malfunctions. The sodium pore normally uncoils to allow sodium ions to flow through, then collapses to close. But the different mutations each cause the pore to malfunction, either not opening enough, remaining open too long or not closing completely, so that sodium ions continue to trickle into the cell, like a drip through a leaking faucet, disrupting normal excitation-contraction coupling of the muscle fibers.
Pure Paramyotonia: Consists of paramyotonia plus (in some but not all patients) episodic weakness which develops with exercise and chilling. Mild attacks cause stiffness, more severe ones cause weakness. Symptoms are worse in the face, neck and upper body. Patients may also have Hyperkalemic Periodic Paralysis (HyperKPP). Some patients are worse after eating food rich in potassium. There’s also a form of PMC without cold sensitivity:
Paramyotonia Congenita von Eulenburg (PMC vE): The symptoms are the same as PMC but the weakness in PMC vE is triggered by hypokalemia (low serum potassium).
Potassium aggravated myotonias (PAM): The PAMs include attacks of paramyotonia which are triggered by potassium. None of these syndromes include weakness. The PAMs are:
Myotonia fluctuans; Mild muscle stiffness that varies in severity from day to day. The myotonia develops during rest within an hour after exercise; lasts for about an hour, worsens with potassium rich food, or and may be severe enough to interfere with breathing. There is no weakness or cold sensitivity.
Myotonia permanens; Severe continuous myotonia (may interfere with respiration) Marked muscular development (hypertrophy), patients look like body-builders but are weak.
Acetazolamide responsive myotonia; Paradoxical myotonia, muscle hypertrophy and muscle pain. Symptoms improve with acetazolamide.
Therapy and Treatment
PMC and PAMs are made worse by:
1. Beta 2-agonists: Fenoterol; Ritodrine
2. Monocarboxylic amino acids
3. Depolarizing muscle relaxants: Succinylcholine, aka diacetylcholine or suxamethonium Each patient has to be treated on an individual basis.
Patients with hyperKPP episodes need to be treated with proven therapies for HyperKPP, with the warning that acetazolomide provokes weakness in a large number of PMC patients. However there are PMC patients who benefit from acetazolomide, and some who take it in an on/off pattern with benefit. Care needs to be taken not to treat hyperkalemic episodes aggressively enough to cause hypokalemia which may trigger a PMC episode.
In some patients with HyperKPP the potassium level overcorrects (drops below normal) at the end of episodes, triggering PMC attacks. Controlling the HyperKPP often helps control the PMC in such patients. Potassium moves into skeletal muscle during episodes of PMC, a shift which also occurs during episodes of Hypokalemic Periodic Paralysis. Serum potassium levels drop during this time and while it may seem contradictory PMC patients may require supplemental potassium to prevent episodes. This therapy must be introduced with caution, and is best done with water soluble potassium which can be mixed in weak solution, but experience has proven that a dose of 5-10 mEq of potassium is often enough to stop an episode of PMC weakness without triggering hyperkalemia.
Paramyotonia may also be treated with the heart drug Mexiletine. We have had numerous reports that low doses (10-20 mg daily) of paroxetine (Paxil) reduce myotonia in PMC, especially in combination with Mexiletine, and may allow for smaller doses of Mexiletine.
References
1. Lajoie, W.J. : Paramyotonia congenita, clinical features and electromyographic findings. Arch. Phys. Med. 42: 507-512, 1961. PMID: 13758355
2. Hudson, A.J. :Progressive neurological disorder and myotonia congenita associated with paramyotonia. Brain 86: 811-826, 1963. PMID: 14090531
3. Samaha, F.J. : Von Eulenburg’s paramyotonia. Trans. Am. Neurol. Assoc. 89: 87-91, 1964. PMID : 5828532
4. McClatchey, A.I.; et al. : Temperature-sensitive mutations in the III-IV cytoplasmic loop region of the skeletal muscle sodium channel gene in paramyotonia congenita. Cell 68: 769-774, 1992. PMID: 1310898
5. Lehmann-Horn, F.; et al.: Adynamia episodica hereditaria with myotonia: a non-inactivating sodium current and the effect of extracellular pH. Muscle Nerve 10: 363-374, 1987. PMID: 3587272
6. Lehmann-Horn, F.; Rudel, R.; Ricker, K.: Non-dystrophic myotonias and periodic paralyses. Neuromusc. Disord. 3: 161-168, 1993. PMID: 7689382
7. Brooke M. H.; Disorders of Skeletal Muscle. Neurology in Clinical Practice, Third Ed, 2000: Bradley, W.G. et al. Eds. Boston, MA: Butterworth/Heinemann.
8. Kimura J. Electrodiagnosis in Diseases of Nerve and Muscle: Principles and Practices. Philadelphia, PA: FA Davis, 1983
9. Subramony SH, Malhotra CP, Mishra SK. : Distinguishing paramyotonia congenita and myotonia congenita by electromyography. Muscle Nerve 1983;6:374-379. PMID: 6888415
10. Paramyotonia Entry Neuromuscular Disease Center, Washington Univ school of Medicine, St. Louis, Mo.
11. Riggs J.E. ; Neurology Clinics; Muscle Disease; The Periodic Paralyses: Vol. 6, No 3 Aug 1988 pp. 485-493
12. Benstead T.J.; Camfield P.R.; King D.B.; Can J Neurol Sci, 1987 May, 14:2, 156-8) Moxley RT; et al. : Neurology, 1989 Jul, 39:7, 952-5) PMID: 2500620
13. Jackson CE, Barohn RJ, Ptacek LJ. : Paramyotonia congenita: Abnormal short exercise test, and improvement after mexiletine therapy. Muscle Nerve 1994;17:763 PMID: 8008003
reviewed 5 October 2020