
Physician’s Summary
Clinical Signs
Hyperkalemic Periodic Paralysis is a genetically determined disorder which causes episodes of weakness and flaccid paralysis of the limbs. The muscles of the eyes, throat, and trunk may also be affected. Episodes last from minutes to hours, very rarely up to 1-2 days. Some patients experience only a few episodes over their lifetime, others have one or more every day. Diagnosis is suggested by episodes of weakness during which the patient’s serum potassium is above 5.0 mmol/l or rises at least 1.0 of a mmol/l above their normal level when not having an episode. About half of patients with HyperKPP also have Paramyotonia Congenita which causes muscle stiffness between attacks.
During episodes sodium moves into the muscles, which may cause a fall in serum sodium levels. Water also moves into the cell causing further hyperkalemia. Hyperregulation may occur at the end of an attack causing hypokalemia, which may result in a misdiagnosis of hypokalemic PP. At the end of an episode the patient may produce a large amount of dilute urine containing increased creatine and experience an increase in serum CK. Between episodes the serum potassium is within normal limits and strength returns to the patient’s normal, but there may be mild muscle stiffness.
Symptoms usually develop in the first years of life, but invariably begin by age 20. Episodes often occur in the early morning following exercise or a meal of potassium-rich food the previous day, especially if the patient has been chilled or is under stress. Episodes occur infrequently to begin with, become more frequent in the 30s and in some patients cease by the time the patient is in their 50s, sometimes to be replaced by permanent muscle weakness. Other patients continue to have episodes into their 70s.
Family History:
Autosomal dominant inheritance (Statistically 50% of an affected person’s offspring will inherit). Age at onset: Infancy to second decade of life. All who inherit the most common genes develop symptoms (complete penetrance) but severity is very variable.
Genetics:
Hyperkalemic periodic paralysis occurs as a result of a mutation in the alpha subunit of the Sodium Channel – (SCN4A); Gene Map Locus 17q23.1-q25.3; Ten Mutations or sequence variants have been identified. Mutations Thr704Met, Met1592Val and Ile693Thr are responsible for 98% of identified cases to date. Mutations Ala1156Thr; Met1592Val, Phe1490Leu, Met1493Ile, Ile693Thr, Ile1495Phe, Ser246Leu, Thr323Met and Leu689Ile are responsible for the remaining two percent of cases. There is reduced penetrance with mutations A1156T & M1360V.
Mutations Arg1448Ser, Arg1448Cys, Arg1448His, and Arg1448Pro are found in HyperKPP patients who also have Paramyotonia Congenita. Met1360Val caused PMC in one family member and HyperKPP in other family members. Thr704Met and Met1592Val have also been identified as causing PMC in single families.
Genetic-Clinical Correlations:
Patients with mutation Thr704Met often develop permanent weakness. Patients with Met1592Val have myotonia; Ala1156Thr & Met1360Val: Reduced penetrance (unaffected carriers can pass gene to next generation.)
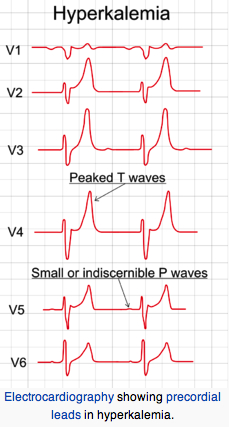
Cardiac Signs and Emergency Treatment of an Attack
For severe hyperkalemia or weakness – inpatient with cardiac monitoring. Mild hyperkalemia or weakness – outpatient with close follow up. Be alert to possible complications: Cardiac arrhythmias, respiratory collapse. May need respiratory support.
Institute emergency treatment if EKG changes occur or serum K > = 7 mEq/L. EKG change sequence: tall peaked T waves, diminished R waves amplitude, increased QRS or PR intervals and P wave disappearance. Slow infusion (over 5 min) of 10% calcium gluconate (reduces cardiac sensitivity to hyperkalemia). Sodium bicarbonate i.v. infusion to raise pH (lower H+) thereby lowering K+ via intracellular shift of H+ and K+ ions (H+ and K+ go in same direction). Infusion of 1 mg/kg D50W containing regular insulin 0.5- 1.0 unit/ml (drives K+ into cells).
Beta-2 adrenergic receptor agonists promote the translocation of potassium into the cell by activation of the Na-K ATPase pump. Salbutamol and other beta-agonists are equally effective given intravenously or via a nebulizer.
The effect of intravenous or nebulized salbutamol is dose-dependent, with an onset of action within 30 min, and a peak effect within 60 min. Nebulized salbutamol is given in doses of 10 or 20 mg. It decreases serum potassium by approximately 1 mmol/L and lasts for at least 2 hours.
Patients’ Descriptions of Hyperkalemic Symptoms & Episodes Typically Include:
Weakness or paralytic episodes which may come on very rapidly. Falls may occur in sudden episodes. Rest after activity is a common trigger. Changes in the daily level of activity and periods of inactivity (sleep, sitting through a movie, car or plane trip, especially in cool temperatures) triggers weakness in many patients. Being outside on a cool windy day can bring on facial tightness and a characteristic “grimace”, with an inability to smile and narrowed eyes.

Photo is of a 65 year-old man with HyperKPP/PMC von Eulenburg outside on a cool and windy November day, trying to smile.
Many patients learn to move constantly to avoid weakness, i.e. rock, ‘fidget’, move their feet. Some patients find that chewing gum is enough ‘activity’ to stave off weakness.
Patients may describe muscle stiffness (myotonia) which can be ‘worked’ or ‘walked’ off. Myotonia is often dismissed as rheumatism, arthritis or fibromyalgia. High carbohydrate food (candy bar) or drink (sugar cola, sweet tea) taken at the first sign of weakness may abort or relieve episodes.
HyperKPP may be accompanied by Paramyotonia Congenita von Eulenburg. This may mean that the patient’s serum potassium is labile during attacks, and paradoxical myotonia may be a prominent feature of attacks. Paradoxical myotonia is myotonia which worsens with activity or exercise, with little to no electrical evidence of myotonia. Aggressive lowering of potassium may precipitate attacks of paramyotonia.
Diagnosis of HyperKPP Genetic Testing
Genetic Testing for identified mutations is available through a number of commercial laboratories, but not all mutations have been identified yet, so a negative DNA test does not rule out HyperKPP.
Compound Muscle Amplitude Test: Positive in 80% of HyperKPP patients
The CMAP (i.e. McManus or Fornier Protocol) test is easier on patients than provocative tests and is almost specific for periodic paralysis. The test is conducted by measuring the CMAP in hypothenar muscle by means of surface electrodes. The test takes approximately 45 minutes and is non-invasive.
Video showing demonstration of CMAP test. (link)
Patient must absolutely be medication-free, i.e. no K+, K+ wasting diuretics, CAI inhibitors, mexiletine, etc. for 72 hours prior to test. CMAP amplitude: Increased immediately after sustained (5 min) maximal contraction Progressively reduced (by 40%) during rest 20 to 40 min after initial increment In Normals: Mildly increased CMAP amplitude after exercise
Provocative Testing: Oral K+ load only if renal, cardiac function and serum K+ are normal (never if arrhythmic). Perform fasting, in am, after exercise 0.05 g/kg KCl in sugar-free liquid over 3 minutes. If negative may give 0.10 to 0.15 g/kg KCl, monitor electrolytes, EKG & strength every 20 minutes. Weakness is typically most noticable after 90 to 180 minutes. IV glucose loading does not provoke weakness in HyperKPP but K+ loading tests (0.05 to .15 g/kg) will induce weakness. K+ loading tests are potentially hazardous and are contraindicated in patients with renal disease and diabetes. Patients should be screened for Andersen-Tawil Syndrome before provocative testing begins. Random serum K+ measurements may suggest the diagnosis, since K+ elevations are frequent during attack-free intervals. EMG evidence of myotonia and finding of vacuoles on muscle biopsy provide supporting data.
Management and Therapy of HyperKPP
The goals of treatment are relief of acute symptoms and prevention of further attacks. Attacks of HyperKPP are brief and seldom severe enough to require emergency treatment. However, progressive permanent weakness can occur with repeated attacks, so treatment should occur as soon as possible.
To Treat Weakness in HyperKPP
Hydrochlorothiazide (25 to 75 mg/day) Acetazolamide (125 to 1,000 mg/d) Dichlorphenamide (50 to 150 mg/day) Albuterol by inhaler or motor-driven nebulizer; Calcium gluconate syrup 15 ml dissolved in sweet tea may serve to abort developing attacks. It is often advisable to prevent hyperkalemic attacks of weakness by the continuous use of a thiazide diuretic or acetazolamide. Thiazide diuretics are preferable because of the possible complications of acetazolamide therapy. The dosage should be kept as low as possible (e.g., 25 mg hydrochlorothiazide daily or every other day). In severe cases, 50 mg or 75 mg of hydrochlorothiazide should be taken daily very early in the morning. Individuals should be monitored so that the serum potassium concentration does not fall below 3.3 mmol/L or the serum sodium concentration below 135 mmol/L
To Treat Muscle stiffness:
Mexiletine 100 to 1,000 mg/day; Paxil 20-40 mg day;
To Treat Paramyotonia ( PMC):
Mexiletine 100 to 1,000 mg/day, Paxil 20-40 mg daily, Potassium supplementation as required 5-10 mEq.
Diet
A high-carbohydrate diet, which avoids food rich in potassium, may be recommended. Glucose or other carbohydrates given during an attack may reduce the severity. Patients can be counselled to drink a sweetened beverage at the first sign of an attack.
Other Considerations Andersen-Tawil Syndrome
Andersen’s Syndrome is a distinct periodic paralysis occurring in the setting of either hyper- or hypokalemia, with severe cardiac involvement (LQT) and skeletal abnormalities. Every patient with periodic paralysis must be screened for this potentially lethal condition. Partial manifestations are common and the subtle nature of the cardiac and dysmorphic features may delay diagnosis but clinical recognition of this syndrome is vital given the predisposition for dysrhythmias and sudden death. Cardiac evaluations using serial ECGs with measurements of the QTc interval are essential and should be performed on all patients undergoing workup for periodic paralysis before provocative tests are performed.
Malignant Hyperthermia in Hyperkalemic PP
Periodic Paralysis patients are at increased risk to Malignant Hyperthermia. During surgery, avoid use of depolarizing anesthetic agents including suxamethonium and anticholinesterases that increase myotonia, which can result in masseter spasm and stiffness of respiratory and other skeletal muscles, and may interfere with intubation and mechanical ventilation.
References:
Hyperkalemic Periodic Paralysis; National Library of Medicine; Frank Weber, MD, PhD
1. Weber F, Jurkat-Rott, K, and Lehmann-Horn F: Gene Reviews; Hyperkalemic Periodic Paralysis , January 28, 2016.
2. Charles G, Zheng C, Lehmann-Horn F, Jurkat-Rott K, Levitt J. Characterization of hyperkalemic periodic paralysis: a survey of genetically diagnosed individuals. J Neurol. 2013;260:2606–13. PMID: 23884711
3. Klingler W, Lehmann-Horn F, Jurkat-Rott K. Complications of anaesthesia in neuromuscular disorders. Neuromuscul Disord. 2005;15:195–206. PMID: 15725581
4. Bendahhou S; Cummins TR; Hahn AF; et al; A double mutation in families with periodic paralysis defines new aspects of sodium channel slow inactivation. J Clin Invest 106:431-8, 2000 PMID: 10930446
5. Sterns RH, Narins RG: Disorders of potassium balance. In: Stein JH, ed. Internal Medicine. 4th Ed. Boston, Little Brown & Co, 1994
6. Hoffman EP: Voltage-gated ion channelopathies: inherited disorders caused by abnormal sodium, chloride, and calcium regulation in skeletal muscle. Annual Review of Medicine 1995;46:431-441 PMID: 7598476
7. Lehmann-Horn F, Rudel R, Jurkat-Rott K. Non-dystrophic myotonias and periodic paralyses. In: Engel AG, Franzini-Armstrong C, eds. Myology: Basic and Clinical. 3 ed. New York: McGraw-Hill; 2004:1257-300.
8. Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev. 1999 Oct;79(4):1317-72. PMID: 10508236.
9. Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F. Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle. Ann Neurol. 1996;39:599–608. PMID: 8619545
10. Mendell, Griggs, Ptacek in Harrison’s ‘Principles of Internal Medicine’ 14th ed. 1998, Chap 14 Levitt, L. P.; Rose, L. I.; Dawson, D. M. : Hypokalemic periodic paralysis with arrhythmia. New Eng. J. Med. 286: 253-254, 1972
11. Sansone, V.; Griggs, R. C.; Meola, G.; et al: Andersen’s syndrome: a distinct periodic paralysis. Ann. Neurol. 42: 305-312, 1999 PMID: 9307251
12. Kuntzer T, Flocard F, Vial C, Kohler A, Magistris M; et al: Exercise test in muscle channelopathies and other muscle disorders. Muscle Nerve 2000 Jul;23(7):1089-94: PMID: 10883004
13. McManis PG; Lambert EH; Daube JR: The exercise test in periodic paralysis. Muscle Nerve, 1986 Oct, 9:8, 704-10 PMID: 3785281
14. Katz JS; Wolfe GI; Iannaccone, S; et al: The Exercise Test in Andersen Syndrome; Archives of Neurology / volume: 56 (page: 352) PMID: 10190827
15. Abbott GW; Butler MH; Bendahhou S; et al; MiRP2 forms potassium channels in skeletal muscle with Kv3.4 and is associated with periodic paralysis. Cell 104:217-31, 2001 PMID: 11207363
16. Kuzmenkin A, Jurkat-Rott K, Lehmann-Horn F, Mitrovic N.; Impaired slow inactivation due to a polymorphism and substitutions of Ser-906 in the II-III loop of the human Nav1.4 channel. Pflugers Arch. 2003 Oct;447(1):71-7. Epub 2003 Jul 26. PMID: 12898257
17. Okuda S, Kanda F, Nishimoto K, Sasaki R, Chihara K. Hyperkalemic periodic paralysis and paramyotonia congenita–a novel sodium channel mutation. J Neurol. 2001 Nov;248(11):1003-4. PMID: 11757950
reviewed and updated May 2022